

PNNL发表开源蛋白质组学软件Informed-Proteomics
分析测试百科网讯 一则消息可能对蛋白组学研究者们最头痛的事有一定的缓解。美国一机构介绍了一款用于分析鉴定完整蛋白质的新型开源软件。
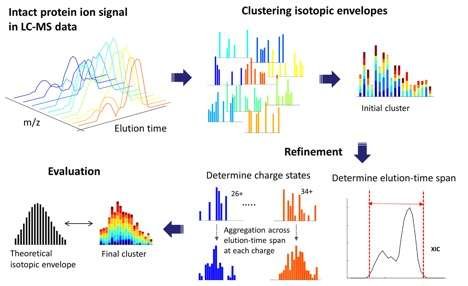
ProMex中的LS-MS特征查找,该功能是Informed-Proteomics软件套件的第一个组件的特征检测算法。 它被开发用于检测完整蛋白质离子的同位素包膜,并正确确定其单同位素质量和丰度
虽然学界们认为,表征蛋白质组对于了解介导疾病的诊断,治疗和预防的蛋白质的活性和功能至关重要,但数量庞大,其结构比基因组更难以表征的蛋白质研究,一度处于停滞状态。
通常,通过液相色谱-质谱(LC-MS)分析策略收集用于表征蛋白质组学的蛋白质组学数据。这样的仪器旨在通过精确测量电荷,质量和重量来显示蛋白质的功能和活性。
主要作者Jungkap Park和太平洋西北国家实验室(PNNL)的科学家们发表了一篇新的Nature Methods论文,介绍了Informed-Proteomics,一种用于通过质谱分析鉴定完整蛋白质的新型开源软件套件。它包含一整套用于自上而下蛋白质组学的新型软件工具,用于分析完整蛋白质。
通过提供新的LC-MS特征查找算法、新的数据库搜索算法、半自动学习方法和交互式结果查看器,高效和精简的信息,软件对蛋白质组学提供了比现有方法更大的改进。
研究蛋白质的“本机结构”
在传统的“自下而上”蛋白质组学方法中,蛋白质被消化成肽用于质谱鉴定。这种方法提供更高的产量,但结果对于完整和活性的蛋白质形式可能是不确定的。
自上而下的方法分析每个蛋白质,而分子是完整的。这样,自上而下的蛋白质组学保留了关于翻译后修饰、同工型和统称为蛋白质形式的分子组合的有价值的信息。
PNNL Integrative Omics科学家团队负责人Sam Payne说,“研究蛋白质本身结构非常重要,因为蛋白质的更多信息得到了保留。不过,他补充说:“研究蛋白质整体有非常独特的挑战。”
Payne表示,自上而下的蛋白质组学的技术障碍是“达到你想要的规模”。从自上而下的方法衍生的谱要复杂得多,需要新的软件工具和新颖的算法来满足他所谓的测量细胞中所有蛋白质的“极具挑战性”的想法。
Payne说:“自上而下,你所期待的是非常大的,而且需要正确的数学来组织一种有效的搜索方式。”
“搜索空间”和乳腺癌检测
为什么这么大的规模?一方面,在自上而下的蛋白质组学中,完整蛋白质的大小意味着电离后的信号分散在许多维度上。另一方面,Payne称之为潜在蛋白模式的“搜索空间”非常大。蛋白质的组合世界可以达数十亿。
作者通过使用已知具有明显差异的人-小鼠异种移植物管腔和基底乳腺肿瘤样品,评估了知情蛋白质组学(Informed-Proteomics)与其他几种常用自上而下蛋白质组学工具。
在分析两种乳腺癌亚型中的3,000种蛋白质形式时,PNNL作者发现,与使用不同方法的最新自上而下分析相比,他们的新软件工具发现了十倍以上差异表达的蛋白质形式。
佩尼表示,PNNL作者的一个优势来自PNNL在“仪器和信息学领域的自上而下分析领域的悠久历史”,这反映了合著者理查德·史密斯(Richard D. Smith)的作品。 “作为一个团队,我们可以在计算和技术分析的各个方面进行改进。”
目前,液相色谱和质谱仪的数据质量一直在不断提高,同时还有样品处理方案的质量。由于采用了更为复杂的自上而下质谱来处理,本文的作者报告说“迫切需要开发可靠的蛋白质形态识别和定量的算法和软件工具。”